PiHKAL: The Chemical Story 7

#76 EMM; 4,5-DIMETHOXY-2-ETHOXYAMPHETAMINE
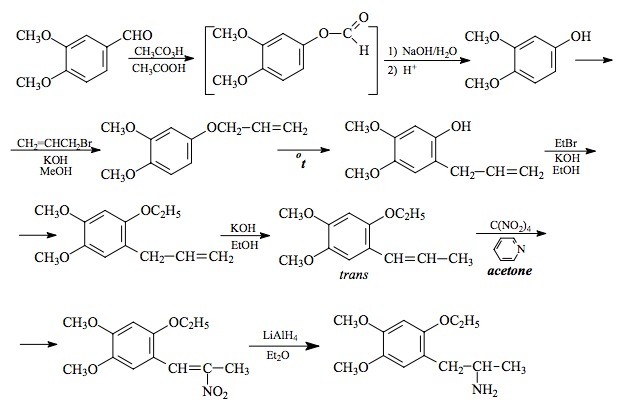

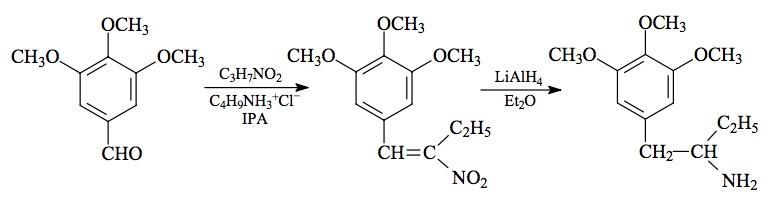
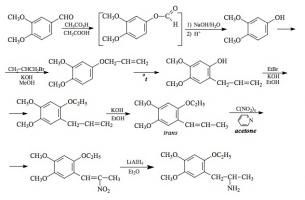
SYNTHESIS: A solution of 166 g 3,4-dimethoxybenzaldehyde in 600 mL acetic acid was well stirred, and brought up to an internal temperature of exactly 25 °C. There was added, in very small portions, a 40% solution of peracetic acid in acetic acid. The evolved heat was removed with an external ice bath, and the rate of addition was dictated by the requirement that the internal temperature should not exceed 25 °C. A total of 210 g of the 40% peracetic acid was used. The reaction mixture was poured into 3 L H2O, and the acetic acid neutralized by the addition of solid K2CO3. The neutral aqueousphase was extracted with 5x150 mL Et2O, and the solvent from the pooled extracts was removed under vacuum. To the red-colored residue there was added 300 mL 10% NaOH, and the mixture was heated for 1 h on the steam bath. This was cooled, washed once with CH2Cl2, acidified with HCl, and extracted with 5x150 mL Et2O. The pooled extracts were washed once with saturated NaHCO3 (which removed most of the color) and the removal of the solvent under vacuum gave 105 g of 3,4-dimethoxyphenol as an amber oil that slowly set up to crystals.
The above crude 3,4-dimethoxyphenol was dissolved in 200 mL EtOH, and treated with a solution of 38.1 g KOH in 300 mL hot EtOH. The clear solution of the potassium salt was a deep red color, and was promptly treated with 94.3 g allyl bromide, at a rate commensurate with the exothermic reaction. The mixture was held at reflux for 2 h. This was then added to 1 L H2O and extracted with 5x100 mL Et2O. The extracts were pooled, and removal of the solvent under vacuum gave a residue of 98 g of a black oil. This was distilled at 104-108 °C at 0.7-1.0 mm/Hg to give 59.3 g 1-allyloxy-3,4-dimethoxybenzene as a pale yellow oil with a greenish cast.
A total of 59 g of the neat 1-allyloxy-3,4-dimethoxybenzene was provided with an internal thermometer, and heated with an open flame. The color quickly became purple, then lightened to a red at 70 °C, and finally to a pale pink by 210 °C. At 240 °C an exothermic reaction set in with the temperature going up to almost 290 °C. It was held in the 270-280 °C range for several min, then allowed to return to room temperature. GC analysis showed two peaks, the second and major one being the desired 1,2,4,5-isomer. A small sample was caught by prep-GC, and it successfully seeded the crude Claissen rearrangement product. The isolated 2-allyl-4,5-dimethoxyphenol, pressed on a porous plate, had a mp of 39.5-40.5 °C which was improved to 41.5-42 °C by recrystallization from hexane.
To a solution of 9.7 g 2-allyl-4,5-dimethoxyphenol in a few mL EtOH, there was added a solution of 2.8 g KOH in 25 mL boiling EtOH followed by 5.5 g ethyl bromide. The mixture was held at reflux for 3.5 h and then poured into 200 mL H2O and extracted with 3x100 mL CH2Cl2. Pooling the extracts and removal of the solvent under vacuum gave a residue of 10.4 g of 4,5-dimethoxy-2-ethoxy-1-allylbenzene as a clear, mobile oil. It was substantially a single component by GC and was used in the following isomerization step without further purification.
A solution of 9.4 g 4,5-dimethoxy-2-ethoxy-1-allylbenzene in 10 mL EtOH was treated with 20 g flaked KOH, and heated on the steam bath. The progress of the isomerization was followed by the assay of isolates by GC. After 5 h, the reaction mixture was poured into 250 mL H2O which immediately generated a pasty solid. This was sucked free of solvent and other liquids on a sintered funnel, giving 5.5 g of trans-4,5-dimethoxy-2-ethoxy-1-propenylbenzene as an amber solid with a mp of 65-67 °C. A small analytical sample from hexane had a mp of 68 °C.
A solution of 5.0 g trans-4,5-dimethoxy-2-ethoxy-1-propenylbenzene in 27 g acetone that contained 2.2 g pyridine was magnetically stirred and cooled to 0 °C. There was then added 4.5 g tetranitromethane and, after 2 minutes stirring at this temperature, the reaction mixture was quenched with a solution of 1.5 g KOH in 26 mL H2O. The reaction mixture remained a clear deep orange color, and additional H2O was required to institute crystallization. There was the slow deposition of bright yellow crystals of 1-(4,5-dimethoxy-2-ethoxyphenyl)-2-nitro-propene which weighed, after EtOH washing and air drying to constant weight of 4.4 g. The mp was 75-76 °C.
To a gently refluxing suspension of 3.5 g LAH in 250 mL anhydrous Et2O under a He atmosphere, there was added 3.9 g 1-(4,5-dimethoxy-2-ethoxyphenyl)-2-nitropropene by allowing the condensing Et2O to drip into a shunted Soxhlet apparatus with the thimble containing the nitrostyrene. This effectively added a warm saturated solution of the nitrostyrene dropwise; the nitrostyrene was very soluble in Et2O. Refluxing was maintained for 2.5 h and the reaction continued to stir at room temperature for an additional 3.5 h. The excess hydride was destroyed by the cautious addition of 225 mL 1.5 N H2SO4. When the aqeous and Et2O layers were finally clear, they were separated, and 75 g of potassium sodium tartrate was dissolved in the aqueous fraction. Aqueous NaOH was then added until the pH was >9, and this was then extracted with 3x100 mL CH2Cl2. Evaporation of the solvent under vacuum produced 2.8 g of a clear, almost colorless oil that was dissolved in anhydrous Et2O and saturated with anhydrous HCl gas. This initially generated a solid that then oiled out. After a few minutes stirring, this began to solidify again and it finally transformed into a loose fine white solid. This was recrystallized by dissolution in 50 mL warm IPA followed by dilution with 300 mL Et2O. After a few minutes, crystals of 4,5-dimethoxy-2-ethoxyamphetamine hydrochloride (EMM) formed which were removed by filtration, Et2O washed, and air dried. These weighed 2.7 g and had a mp of 171-172 °C. Anal. (C13H22ClNO3) C,H,N.
DOSAGE: greater than 50 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 50 mg) There were no effects.
EXTENSIONS AND COMMENTARY: This was the first of the ethoxy homologues of TMA-2, and it was immediately (well, within a couple of months) run up from an initial dab to 25 milligrams. This was in early 1963, and the lack of activity of EMM was keenly disappointing. This was a level at which the prototype, TMA-2, was very active, and the conclusion was that maybe any change on the molecule would result in a loss of activity. So this approach was shelved for a while, and all efforts were directed into the relocation, rather than the elongation, of the methoxy groups. A few months later, the ethoxy question was addressed again, and the discovery of MEM rekindled full interest in this ethoxy question.
#77 ETHYL-J; 2-ETHYLAMINO-1-(3,4-METHYLENEDIOXYPHENYL)BUTANE;
N-ETHYL-1-(1,3-BENZODIOXOL-5-YL)-2-BUTANAMINE
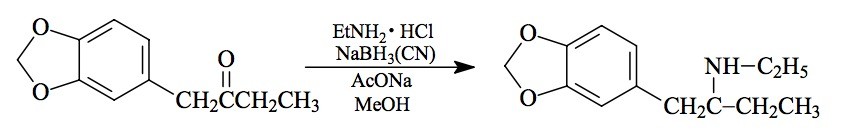

SYNTHESIS: A stirred solution of 9.0 g 1-(3,4-methylenedioxyphenyl)-2-butanone (see the recipe for J for its preparation) in 150 mL MeOH was treated with 9.0 g ethylamine hydrochloride, 4.0 g anhydrous NaOAc, and 3.0 g sodium cyanoborohydride. The pH was maintained between 6 and 7 by the periodic addition of HCl. After the base formation had stabilized, there was added an additional 9.0 g ethylamine hydrochloride, 9.0 g NaOAc and 2.0 g sodium cyanoborohydride. With continuous stirring, there was HCl added over the course of 1 h until the final pH was approximately 2. The reaction mixture was poured into 700 mL dilute NaOH, and extracted with 3x75 mL CH2Cl2. These extracts were pooled, and back-extracted with dilute H2SO4. This was washed with 2x50 mL CH2Cl2, then made basic with dilute NaOH and extracted with 2x75 mL CH2Cl2. Removal of the solvent under vacuum gave a 0.81 g residue which was dissolved in 10 mL IPA. Neutralization with concntrated HCl formed white crystals spontaneously. These were diluted with Et2O, filtered, Et2O washed and air dried to provide 0.85 g 2-ethylamino-1-(3,4-methylenedioxy-phenyl)butane hydrochloride (ETHYL-J), with mp of 176-177 °C. Anal. (C13H20ClNO2) C,H. The neutral fraction that remained in the organic phase following the dilute sulfuric acid extraction, was recovered by removal of the solvent under vacuum. There was obtained about 5 g of an amber liquid that was largely 2-hydroxy-1-(3,4-methylenedioxyphenyl)butane.
DOSAGE: greater than 90 mg.
DURATION: probably short.
QUALITATIVE COMMENTS: (with 65 mg) Perhaps aware at 20 minutes. Definitely aware at 45 minutes. Diffusing to nothing at 3-4 hours.
(with 90 mg) I am somewhere between 1 and +. And everything became lost in the evening with a couple of glasses of wine and talk that went on to 3 AM.
EXTENSIONS AND COMMENTARY: And nothing higher has ever been looked at. If the analogy with the amphetamine counterparts (J with MDA, METHYL-J with MDMA, and this, with MDE) were to hold up (a drop of about a third in potency with the lengthening of the chain by a carbon atom), one might guess that this compound would be an interesting intoxicant, but probably not until you got up into the area at or above a 200 milligram dose. And that is a lot of chemical for the body to have to handle. Some day, maybe.
#78 ETHYL-K; 2-ETHYLAMINO-1-(3,4-METHYLENEDIOXYPHENYL)PENTANE;
N-ETHYL-1-(1,3-BENZODIOXOL-5-YL)-2-PENTYLAMINE
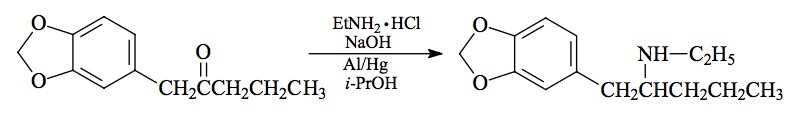

SYNTHESIS: A solution of 120 mg mercuric chloride in 160 mL H2O was poured over 4.7 g aluminum foil (Reynolds Wrap, regular weight, cut into 1 inch squares) and allowed to stand until the amalgamation was well underway (about 30 min). The H2O was then drained and the foil washed with 2x200 mL H2O with thorough draining. There was then added, in sequence and with good swirling and agitation between each addition, 8.5 g ethylamine hydrochloride dissolved in 7 mL H2O, 21 mL IPA, 17 mL 25% NaOH, 7.1 g 1-(3,4-methylenedioxyphenyl)-2-pentanone (see the recipe for METHYL-K for its preparation), and finally 40 mL IPA. The reaction mixture was periodically heated on the steam bath to keep the reaction moving and active. After all the metal had been consumed, the mixture was filtered, and the filter cake washed with MeOH. The solvent was removed from the combined filtrate and washings, and the residue suspended in 800 mL dilute HCl. This was washed with 3x100 mL Et2O, made basic with 25% NaOH, and extracted with 3x100 mL CH2Cl2. The pooled extracts were stripped of solvent under vacuum yielding a residue of 6.3 g of an amber oil. This was distilled at 115-125 °C at 0.4 mm/Hg to give 5.61 g of an almost white liquid which was dissolved in 28 mL IPA, neutralized with concentrated HCl, and diluted with 100 mL anhydrous Et2O. The resulting clear solution became cloudy, then set up in a cottage cheese texture, and then all broke up to a beautiful loose solid. This was filtered, Et2O washed and air dried to give 5.99 g 2-ethylamino-1-(3,4-methylenedioxyphenyl)pentane hydrochloride (ETHYL-K) with a mp of 157-158 °C. Anal. (C14H22ClNO2) C,H.
DOSAGE: (greater than 40 mg).
DURATION: unknown.
QUALITATIVE COMMENTS: (with 40 mg) There was a paresthetic twinge in my shoulder area at about an hour Q other than that, absolutely nothing.
EXTENSIONS AND COMMENTARY: And that is as high a dose as has apparently ever been tried with ETHYL-K. The compounds with the hexane chain (L-series) rather than the pentane chain of the K-series have been made, but they have been spun into the recipe for METHYL-K.
#79 F-2; 2-M; 6-(2-AMINOPROPYL)-5-METHOXY-2-METHYL-2,3-DIHYDROBENZOFURAN
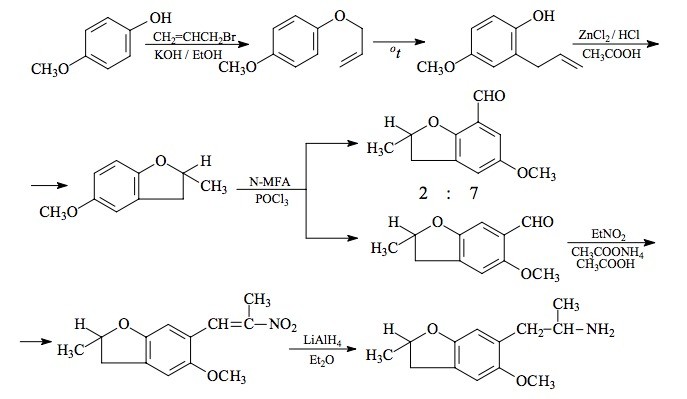

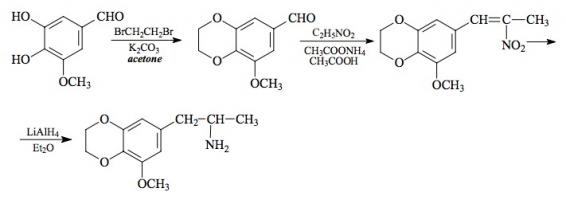
SYNTHESIS: To a solution of 43.2 g KOH pellets in 250 boiling EtOH there was added 96 g 4-methoxyphenol followed by the slow addition of 131.2 g allyl bromide, and the mixture was held under refluxing conditions for 16 h. After cooling, the reaction was added to 1.6 L H2O, and made strongly basic with 25% NaOH. This was extracted with 3x100 mL CH2Cl2, the extracts pooled, washed once with dilute NaOH and then once with dilute HCl. Removal of the solvent under vacuum gave 93.8 g of 4-allyloxyanisole as a pale amber oil, which was used in the following reaction without further purification.
A round-bottomed flask containing 93 g crude 4-allyloxyanisole was equipped with an immersed thermometer and heated with an external flame until an exothermic reaction set in at 230 °C. The temperature rose to 270 °C and it was maintained there with the flame for five minutes. After cooling to room temperature, the reaction mix was poured into 2 L H2O and made strongly basic with the addition of 25% NaOH. This dark aqueous phase was washed with 2x200 mL CH2Cl2, and then acidified with HCl. This was then extracted with 2x200 mL CH2Cl2, and the pooled extracts washed first with saturated NaHCO3 and then with H2O. Removal of the solvent under vacuum gave 65.6 g of 2-allyl-4-methoxyphenol as a clear, amber oil. To a solution of 1.66 g of this crude phenol in 5 mL hexane with just enough CH2Cl2 added to effect a clear solution, there was added 1.3 g phenyl isocyanate followed with three drops of triethylamine. An exothermic reaction ensued which spontaneously deposited white crystals. These was removed and hexane washed to give 2-allyl-4-methoxyphenyl N-phenyl carbamate, with a mp of 88-89 °C. The acetate ester, from the phenol and acetic anhydride in pyridine, did not crystallize.
To a solution of 37.7 g 2-allyl-4-methoxyphenol in 125 mL glacial acetic acid there was added 19 g zinc chloride followed with 63 mL concentrated HCl. The mixture was held at reflux temperature for 40 min, then cooled to room temperature, diluted with 300 mL H2O, and extracted with 2x200 mL CH2Cl2. The pooled extracts were washed repeatedly with 8% NaOH until the washings remained basic. Removal of the solvent under vacuum gave a clear pale yellow oil that was distilled at the water pump. A fraction boiling at 150-165 °C was 5-methoxy-2-methyl-2,3-dihydrobenzofuran which weighed 25 g and which was a highly refractive colorless oil. The infra-red spectrum indicated that some small amount of hydroxy group was present, but the NMR spectrum was in complete accord with the benzofuran structure. A higher cut in this distillation gave 4.5 g of a phenolic product tentatively assigned the structure of 4-methoxy-2-propenylphenol. The target dihydrobenzo-furan has also been synthesized from the open-ring o-allyl phenol in acetic acid solution with the addition of a catalytic amount of concentrated H2SO4.
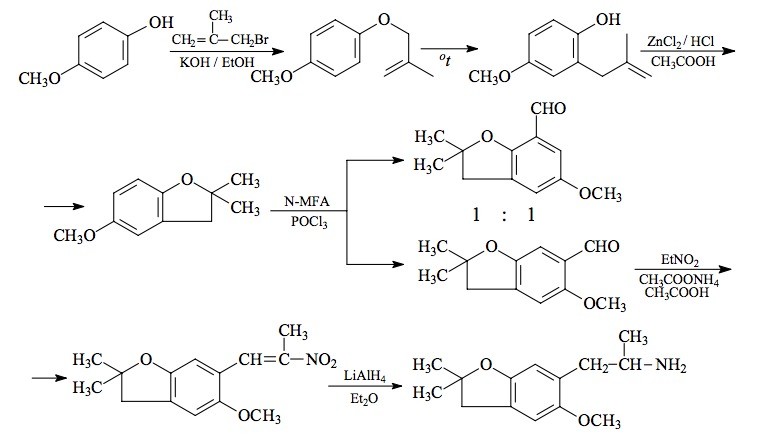
To a half-hour pre-incubated mixture of 69 g POCl3 and 60 g N-methylformanilide there was added 29.0 g 5-methoxy-2-methyl-2,3-dihydrobenzofuran and the mixture was heated on the steam bath for 2 h. The reaction mixture was poured into 1 L H2O, and allowed to stir overnight. The brown gummy solids were removed by filtration, and air dried as completely as possible. These weighed 32 g and were shown by GC on OV-17 to consist of two benzaldehyde isomers in a ratio of 7:2. This was triturated under 18 mL MeOH, and the undissolved solids removed by filtration and washed with 6 mL additional MeOH. The mother liquor and washings were saved. The 17.8 g of dull yellow solids that were obtained were repeatedly extracted with 75 mL portions of boiling hexane (4 extracts were required) and each extract, on cooling, deposited yellow crystals of the major aldehyde. The dried crystals of 6-formyl-5-methoxy-2-methyl-2,3-dihydrobenzofuran were combined (9.5 g) and had a mp of 80-82 °C. The methanol washes saved from above were stripped of solvent, and the sticky, orange solids that remained were enriched in the minor aldehyde isomer (3:2 ratio). Several injections of this crude material into a preparative GC OV-17 column gave sufficient quantities of the "wrong" isomer for NMR characterization. The 2-methyl group was intact (eliminating the possibility of a dihydrobenzopyran isomer) and the ring meta-proton splitting required that the formyl group be in the benzofuran 7-position. This crystalline solid was, therefore, 7-formyl-5-methoxy-2-methyl-2,3-dihydrobenzofuran.
A solution of 9 g of 6-formyl-5-methoxy-2-methyl-2,3-dihydrobenzofuran in 35 mL glacial acetic acid was treated with 6 mL of nitroethane followed with 3.1 g anhydrous ammonium acetate. This mixture was heated on the steam bath for 4 h, diluted with half its volume with warm H2O, and seeded with a bit of product that had been obtained separately. The slightly turbid solution slowly crystallized as it cooled, and was finally held at 0 °C for several h. The deep orange product was removed by filtration, washed with 50% acetic acid, and air dried to constant weight. There was thus obtained 7.0 g 5-methoxy-2-methyl-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran with a mp of 89-90 °C from MeOH.
A suspension of 5.0 g LAH in 500 mL of well stirred anhydrous Et2O at a gentle reflux, was treated with a warm, saturated solution of 7.0 g 5-methoxy-2-methyl-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran in Et2O added dropwise. The mixture was kept at reflux temperature for 36 h, allowed to stand 2 days, and then the excess hydride destroyed by the cautious addition of 500 mL 6% H2SO4. The phases were separated, and the aqueous phase washed with 2x200 mL CH2Cl2. A total of 125 g potassium sodium tartrate was added to the aqueous phase, and sufficient 25% NaOH added to bring the pH to about 10. This phase was extracted with 3x150 mL CH2Cl2, and the pooled extracts were stripped of solvent under vacuum. The residual oil (4.8 g, amber in color) was dissolved in 300 mL anhydrous Et2O which, upon saturation with anhydrous HCl gas gave a clear solution that suddenly deposited white crystals. The hydrochloride salt of 6-(2-aminopropyl)-5-methoxy-2-methyl-2,3-dihydrobenzofuran weighed 2.3 g and was not satisfactory as a solid derivative, but it appears that the oxalate salt is both nonhygroscopic and quite stable. It (F-2) had a mp of 216-218 °C and it displayed a textbook NMR.
DOSAGE: greater than 15 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: This material, which is certainly a mixture of two diastereoisomeric pairs of racemates since there are two chiral centers present, showed no effects at levels of up to 15 milligrams orally. Doses of 100 mg/Kg were without effects in mice following i.p. injections, although half again this amount proved to be lethal. In rats trained to discriminate LSD from saline, F-2 proved to be about 40 times less potent than the reference compound DOM, requiring some 5 mg/Kg for positive responses. But the human trials were only up to about 0.2 mg/Kg.
This was the prototype compound that was originally put together to justify giving a paper at a marijuana conference in Sweden, in 1968. Although I had never done much with marijuana or with its principal ingredients, I thought maybe I could bend the topic a bit to embrace some potentially active phenethylamines. There is a story of an international conference held in Geneva a few years earlier to discuss the worrisome decrease in the elephant population. A German zoologist invested a full eight-hour day in a summary of his 21 volume treatise on the anatomy and the physiology of the elephant. A French sociologist presented a lively slide show on the mating rituals and rutting behavior of the elephant. And a rabbi from Tel Aviv entitled his talk: "Elephants and the Jewish Problem." My Swedish talk should have been named "Marijuana and the Psychedelic Amphetamines." The memorable story of meeting the chief of the Swedish equivalent of the Bureau of Narcotics, and ending up playing Mozart sonatas in the attic of his home, has been spun out elsewhere in the book.
The original concept was a grand plan to imitate two of the three rings of tetrahydrocannabinol. There is an aromatic ring (with an alkyl group and two oxygens on it) and it is fused to a pyran ring with a couple of methyl groups on it. So, if one were to tie the methyl group at the 4-position of DOM around with a short carbon chain into the oxygen atom at the five position, one could squint and say that the resulting amphetamine was kinda something like an analogue of THC. Thus, the resulting six-membered ring (a pyran) or five-membered ring (a furan) could be peppered with methyl groups at different locations (and up to two per location). If the ring was a five-membered structure, then the parent system would be a benzofuran, and the location of methyl groups on the ring would be indicated by the appropriate numbers following the letter RFS which would stand for "furan". And if it were to be a six-membered ring, the resulting benzopyran would be indicated with a RPS for pyran, and again the methyl group or groups would be indicated by the substitution position. This code would cover all polymethylated homologues with codes that would look like F-22 and P-2234. If any of them showed up with fascinating activities, I would extend methyls to ethyls, and work out some whole new naming code at some future time. An early system, naming this compound 2-M for a methyl group on the 2-position of the furan ring, was abandoned when it became apparent that the pyran world would screw everything up.
The isolation of characterizable quantities of 7-formyl-5-methoxy-2-methyl-2,3-dihydrobenzofuran from the benzaldehyde recipe above gave a fleeting fantasy of a whole new direction that this little project might go. If this unexpected benzaldehyde were to be converted to the corresponding amphetamine, one would have 7-(2-aminopropyl)-5-methoxy-2-methyl-2,3-dihydrobenzofuran. Suddenly here would be a 2,3,5-trisubstituted thing with a ring at the 2,3-position, similar to the still unmade MMDA-4. The temptation to be diverted in this way lasted, fortunately, only a few minutes, and the project was shelved. Someday, when there are buckets of spare time or hosts of eager graduate students, some fascinating chemistry might lie this way, and maybe some fascinating pharmacology, even.
The plain furan analogue, without any methyl groups on it, has been made. Five-methoxybenzofuran formed the 6-formyl derivative (the aldehyde) with a mp of 79-80 °C and from it the nitrostyrene (orange needles, mp 89-91 °C) and the final amphetamine (white solids, as the methane sulfonate, mp 141-144 °C) were prepared in a manner similar to the preparation of F-2 above. In the rat studies, it was three times more potent than F-2, but still some 15 times less potent than DOM. And in initial human trials (of up to 30 milligrams) there were again no effects noted. Naming of this material is easy chemically (6-(2-aminopropyl)-5-methoxy-2,3-dihydrobenzofuran) but tricky as to code. If the numbers that follow the RFS give the location of the methyl groups, then this material, without any such groups, can have no numbers following, and should properly be simply RF.S OK, it is RF.S The preparation or the attempted preparations of other homologues such as F-23 and F-233 are outlined under the recipe for F-22.
#80 F-22; 6-(2-AMINOPROPYL)-2,2-DIMETHYL-5-METHOXY-2,3-DIHYDROBENZOFURAN

SYNTHESIS: To a solution of 43.2 g flaked KOH in 250 mL hot EtOH there was added 96 g 4-methoxyphenol followed by 90 g 2-methylallyl chloride over the course of 2 h. The mixture was held at reflux for 24 h, then added to 1.6 L H2O. There was sufficient 25% NaOH added to make the phase strongly basic, and this was then extracted with 3x200 mL CH2Cl2. The pooled extracts were washed with H2O, and the solvent removed under vacuum. The residue, 125 g of a pale amber oil, was crude 4-(2-methylallyloxy)anisole and was used without further purification in the following reaction.
In a round-bottomed flask containing an internal thermometer, there was placed 125 g of unpurified 4-(2-methylallyloxy)anisole, and this was heated with an open flame. At an internal temperature of 190 °C an exothermic reaction set in, raising the temperature to 250 °C, where it was held for an additional 2 min. After the reaction mixture had cooled to room temperature, it was poured into 500 mL H2O, made strongly basic with 25% NaOH, and extracted repeatedly with 100 mL portions of CH2Cl2 until the extracts were essentially colorless. These extracts were pooled and the solvent removed to provide 80.0 g of a deeply colored oil that proved to be largely the appropriately substituted dihydrobenzofuran. The aqueous residue from above was acidified with concentrated HCl, and again extracted with CH2Cl2. Removal of the solvent gave 17.7 g of 4-methoxy-2-(2-methylallyl)phenol as an amber oil which eventually set down as white crystals with a mp of 52.5-54 °C.
A solution of 17 g of 4-methoxy-2-(2-methylallyl)phenol in 56 g acetic acid was treated with 8.4 g zinc chloride followed with 28 mL concentrated HCl. This mixture was heated at reflux temperature with a mantle for 1 h. After cooling, this was poured into H2O and extracted with 2x150 mL CH2Cl2. The pooled extracts were washed with several portions of 8% NaOH, until the extracts were colorless. The organic fraction was then washed with H2O, and the solvent removed to yield 5.8 g of 2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran as a pale amber oil with a pungent smell. This was purified by distillation, giving a fraction of an off-white oil with a bp of 136-138 °C at 33 mm/Hg.
To a mixture of 8.0 g N-methylformanilide and 9.2 g POCl3 which had been allowed to stand for 0.5 h, there was added 4.0 g 2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran, and the mixture held at the steam bath temperature for 2.5 h. This was then poured into 200 mL H2O which produced a black oily phase that gave no hint of crystallization. This mixture was extracted with 3x150 mL CH2Cl2 and the solvent was removed from the pooled extracts under vacuum. The residual oil (which was shown by GC to contain approximately equal quantities of two isomeric benzaldehydes A and B) was extracted with three 75 mL portions of boiling hexane, each of which on cooling deposited a reddish oil that partially crystallized. A fourth hexane extract gave nothing more. The solvent was decanted from these three extracts, and the semi-solid residues were ground under 3.0 mL MeOH giving 1.4 g of pale yellow crystals of 2,2-dimethyl-6-formyl-5-methoxy-2,3-dihydrobenzo-furan, isomer RBS. After recrystallization from MeOH, the color was almost white, and the mp was 79.5-80.5 °C. The combined mother liquors were enriched in isomer RAS which proved, following preparative GC separation and NMR analysis, to be the 7-formyl isomer. The 80 g of impure dihydrobenzofuran isolated from the Claisen rearrangement above was distilled and a fraction (43.8 g) that boiled from 138-153 °C at 30 mm/Hg was processed as described here to the aldehyde mixture. Following similar hexane extractions, a yield of 4.0 g of a 95% pure isomer RBS was finally obtained. The remaining components of this fraction were not determined, but it is possible that there were some that contained the six-membered benzopyran ring system.
To a solution of 5.2 g of 2,2-dimethyl-6-formyl-5-methoxy-2,3-dihydro-benzofuran in 20 mL glacial acetic acid there was added 3 mL nitroethane followed by 1.6 g anhydrous ammonium acetate. This mixture was heated for 4 h on the steam bath, and then a small amount of H2O was added to the hot solution. This instigated the formation of a copious deposition of brick-red crystals which were, after cooling, removed by filtration, and recrystallized from 50 mL boiling MeOH. After air drying there was thus obtained 2.7 g of day-glo yum-yum orange crystals of 2,2-dimethyl-5-methoxy-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran. An additional 0.6 g of product was obtained by working the mother liquors.
A suspension of 2.5 g LAH in 300 mL refluxing anhydrous Et2O was treated with a solution of 3.1 g 2,2-dimethyl-5-methoxy-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran in Et2O. The mixture was held at reflux temperature for 18 h. After cooling, the excess hydride was destroyed by the cautious addition of 400 mL H2O which contained 15 g H2SO4. The aqueous phase was separated, washed once with Et2O, and then once with CH2Cl2. There was then added 60 g potassium sodium tartrate, and the pH was brought to above 10 by the addition of 25% NaOH. This was extracted with 3x250 mL CH2Cl2, the extracts pooled, and the solvent removed under vacuum. There remained 2.8 g of an amber oil with an ammoniacal smell. This was dissolved in 200 mL anhydrous Et2O, and saturated with anhydrous HCl gas. There was the immediate formation of an oil, from which the supernatent Et2O was decanted. The residual oil was resuspended in a second 200 mL anhydrous Et2O, again decanted, and finally a third 200 mL Et2O effected the dissolving of the remaining oil to give a clear solution. All three solutions became gelatinous over the following few h, and each deposited a crop of white crystals over the following few days. From the first there was obtained 1.4 g of product with a mp of 153-154 °C; from the second, 0.2 g with a mp of 153-154 °C; and from the third, 1.2 g with a mp of 155-156 °C. These crops were combined, and recrystallized from 10 mL of boiling CH3CN to give 1.7 g 6-(2-aminopropyl)-2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran hydrochloride (F-22) as a white crystalline solid which had a mp of 154-155 °C. This material, even when dry, showed a tendency to discolor with time.
DOSAGE: greater than 15 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: And here is yet another dihydrobenzofuran which is not of a very high potency if, indeed, it is active at all. This particular dihydrobenzofuran analogue, F-22, had sort of tickled my fancy as being an especially good candidate for activity. It had a certain swing to it. F-22, like LSD-25. And here it was finished, just five days before I had to deliver a paper concerning the syntheses (and activities°) of all these dihydrobenzofurans to the marijuana congress. Could this possibly be another LSD? I was sufficiently convinced that the possibility was real, that I actually started the screening process at a most unusually low level of 10 micrograms. Two days later, I upped this to a dose of 25 micrograms (no activity again) and three days after that, at 1 AM on the polar flight to Copenhagen, I swallowed the "monstrous" dose of 50 micrograms. Shoot the works. If I were to blossom all over the tourist section of the SAS plane, well, it would be quite a paper to give. If not, I could always say something like, "The active level has not yet been found." No activity. Another Walter Mitty fantasy down the tubes.
And, as it turned out, the entire project pretty much ran out of steam. A number of clever analogs had been started, and would have been pursued if there had been any activity promised of any kind with any of these dihydrobenzofurans. The "other" benzaldehyde described above, could have been run in a manner parallel to that proposed for the counterpart with F-2, to make the eventual amphetamine, 7-(2-aminopropyl)-2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran. Great strides had been made towards F-233 (I have discussed the naming system under F-2, with the F standing for the furan of benzofuran and the 2 and 3 and 3 being the positions of the methyl groups on it). The reaction of 4-methoxyphenol with 1-chloro-3-methyl-2-butene gave the ether which underwent the thermal Claisen rearrangement to 2-(1,1-dimethylallyl)-4-methoxyphenol with a bp of 148-157 °C at 30 mm/Hg. This was cyclized to the intermediate cycle 2,3,3-trimethyl-2,3-dihydrobenzofuran which, after distillation, was shown to be only 80% pure by GC analysis. This was, nonetheless, (and with the hope that is in the very fiber of a young innocent chemist), pushed on to the benzaldehyde stage (and there were a not-too-surprising four benzaldehydes to be found in the oil that was produced, which refused to crystallize). And then (when sheer desperation replaced hope) these were condensed with nitroethane to form an even worse mixture. Maybe something might crystallize from it? Nothing ever did. Junk. Everything was simply put on the shelf where it still rests today, and F-233, 6-(2-aminopropyl)-5-methoxy-2,3,3-trimethyl-2,3-dihydrobenzofuran, remains the stuff of speculation.
And a start towards F-23, 6-(2-aminopropyl)-2,3-dimethyl-5-methoxy-2,3-dihydrobenzofuran, got just as far as the starting ether, when it occurred to me that the final product would have an unprecedented three chiral centers, and so a total of four racemic pairs of diastereoisomers. And then I discovered that the starting allyl halide, crotyl chloride, was only 80% pure, with the remaining 20% being 3-chloro-1-butene. This would have eventually produced a 2-ethyl-analogue, 6-(2-aminopropyl)-2-ethyl-5-methoxy-2,3-dihydrobenzofuran, with its two chiral centers and two more pairs of stereoisomers (not to speak of the need to devise an entirely new coding system). Unless something were to fall into my lap as a crystalline intermediate, the final mess could have had at least six discreet compounds in it, not even considering optical isomers. And I haven't even begun to think of making the six-membered dihydrobenzopyrans which were the THC analogues that presented the rationale that started the whole project in the first place. A recent issue of the Journal of Medicinal Chemistry has just presented an article describing the reaction of 6-methoxytetrahydrobenzopyran with dichloromethyl methyl ether, and approximately equal amounts of all three of the possible isomers were obtained. That would have been the first step towards making the prototypic compound 7-(2-aminopropyl) 6-methoxy-1,2,3,4-tetrahydrobenzopyran. Just as the benzofurans were all named as F-compounds, this, as a benzopyran, would have been a P compound, but P also is used for proscaline, and there would have been some repair-work needed for these codes.
Time to abandon ship. The fact that I had just synthesized and discovered the strange activity of ARIADNE at about this time, made the ship abandonment quite a bit easier to accept.
#81 FLEA; N-HYDROXY-N-METHYL-3,4-METHYLENEDIOXYAMPHETAMINE
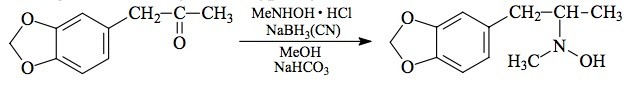

SYNTHESIS: (from 3,4-methylenedioxyphenylacetone) A solution of 2.1 g N-methylhydroxylamine hydrochloride and 4.4 g 3,4-methylenedioxyphenylacetone in 5.5 mL MeOH was added to a suspension of 4.5 g NaHCO3 in 30 mL boiling MeOH. There was added about 5 mL H2O (which gave a clear solution) followed by another 50 mL H2O which produced a pale yellow color. To this solution of the unisolated nitrone there was added 1.7 g sodium cyanoborohydride, which generated a goodly amount of foaming. There was HCl added as needed to maintain the pH at about neutrality. The reaction appeared to have stopped after a day or two, so all was poured into 500 mL H2O, acidified with HCl, and washed with 2x75 mL CH2Cl2. The addition of base brought the pH >9, and this was then extracted with 3x75 mL CH2Cl2. Removal of the solvent from the pooled extracts gave a residue of 1.65 g of crude N-hydroxy-N-methyl-3,4-methylenedioxyamphetamine. Efforts to obtain solid seed samples of the salts with hydrochloric acid, perchloric acid, sulfuric acid, phosphoric acid, and with a number of organic acids, all failed. The salt formation from this free-base will be discussed below.
(from MDOH) A solution of 0.75 g crystalline free-base MDOH in a few mL MeOH was treated with a solution of 0.4 g sodium cyanoborohydride in 10 mL MeOH, and there was then added 2 mL of 35% formaldehyde. The stirred reaction mixture was kept at a neutral pH with the occasional addition of HCl. After several days (when additional acid was no longer required) the excess solvent was removed under vacuum, and the residue poured into dilute H2SO4. This was washed with 2x75 mL CH2Cl2 and then, following the addition of base, this was extracted with 3x75 mL CH2Cl2. Removal of the solvent from the pooled extracts gave a viscous oil residue of 0.53 g. The free-base product from these preparations was distilled at 110-120 °C at 0.2 mm/Hg to give the N-hydroxy-N-methyl product as a white oil. An alternate methylation procedure used a solution of MDOH in a 4:1 MeOH/acetic acid solution containing formaldehyde which was reduced with sodium borohydride at dry ice temperatures. Its work-up is identical to that involving sodium cyanoborohydride.
The distilled product was dissolved in an equal volume of MeOH, and treated with a half-equivalent of oxalic acid dihydrate, dissolved in 10 volumes of MeOH. This combination gave the slow deposition of crystals of the full oxalate salt (one acid, two bases) as a white crystalline product. The mp of the crude salt was in the 130-150 °C range, and after recrystallization from CH3CN, N-hydroxy-N-methyl-3,4-methylenedioxyamphetamine oxalate (FLEA) had a mp of 146-147 °C.
DOSAGE: 100 - 160 mg.
DURATION: 4 - 8 h.
QUALITATIVE COMMENTS: (with 90 mg) The material tastes terrible, like grapefruit juice that has stayed in the can too long. There was no nausea, no feeling of difficulty in swallowing at any time during the day. I felt a dry mouth and was thirsty Q sipped water throughout the day. At the beginning of the experiment, there was a glimmer of the MDMA warmth, but later I felt separated and a bit isolated. I was just floating around, seeing the beauty of colors and objects in the house and outdoors and listening first to this conversation, then to that one. All senses seemed enhanced. I found the material pleasant. I was happy with the amount I took but would not be afraid to take more or to take a supplement. I found it similar to, but not the same as, MDMA.
(with 110 mg) We found this very similar to MDMA, but perhaps slightly slower. I plateau'd at 2:30 hours and had a very gradual descent. My friend had a marvelous and private 'cone of silence' that was to him unique to MDMA or to 2C-T-8. Teeth problems were minor, and the descent from the top of the experience showed less interactive, and more contemplative action, than with MDMA. Very similar to MDMA, but with its own character.
(with 110 mg) The onset was at about a half-hour. The come-on was more gradual and much easier than with MDMA, and it seemed to be more head than body oriented. I had about two hours of very complex and personal self-evaluation, and I am not at peace in putting all of it down here in writing. Overall I like it, and I would be interested to see if there's a difference in conjunction with MDMA. Thanks very much.
(with 110 mg + 35 mg) I saw my onset at 20 minutes, and it was subtle, and very pleasant, and had a mild amphetamine-like elevation for me (body lightness, cognitive functions seemed clear and clean, heightened visual awareness and with some enhancement of color). It seemed as if I were on the fringe of LSD-like visual changes, but that never materialized. The affect was very good, communicative, friendly, accepting, but without the profound emotional bonding of MDMA. The following day felt very much like a post-LSD day; we felt great. The body was light, energy good, emotions high, several insights throughout the day, interactions clear and open Q a magnificent gift of a day. I started a menstrual period the day of the experience and it lasted 6 to 7 days; all of this was a couple of weeks early. I have a very favorable impression of FLEA although the body penalty seems high.
EXTENSIONS AND COMMENTARY: Most people who were involved with the evaluation of FLEA quite logically compared it with MDMA, as it was presented as being a very close analogue which might share some of the latter's properties. And to a large measure, the comparison was favorable. The dosages are almost identical, the chronological course of action is almost identical, and there are distinct similarities in the effects that are produced. If there is a consensus of similarities and differences it would be that it is not quite as enabling in allowing a closeness to be established with others. And perhaps there is more of a move towards introspection. And perhaps a slightly increased degree of discoordination in the thought processes. But also, part of this same consensus was that, were MDMA unknown, this material would have played its role completely.
And from the scientific point of view, it lends more weight to a hypothesis that just might be a tremendous research tool in pharmacology. I first observed the intimate connection between an amine and a hydroxylamine with the discovery that N-hydroxy-MDA (MDOH) was equipotent and of virtually identical activity to the non-hydroxylated counterpart (MDA). And I have speculated in the recipe for MDOH about the possible biological interconversions of these kinds of compounds. And here, the simple addition of a hydroxyl group to the amine nitrogen atom of MDMA produces a new drug that is in most of its properties identical to MDMA. The concept has been extended to 2C-T-2, 2C-T-7, and 2C-T-17, where each of these three active compounds was structurally modified in exactly this way, by the addition of a hydroxyl group to the amine nitrogen atom. The results, HOT-2, HOT-7 and HOT-17 were themselves all active, and compared very closely with their non-hydroxylated prototypes.
Just how general might this concept be, that an N-hydroxyl analog of an active amine shall be of similar action and duration as the parent drug? What if it really were a generality° What havoc it would wreak in the pharmaceutical industry° If I could patent the concept, then I would be able to make parallel best sellers to all of the primary and secondary amines out there in the industry. Perhaps 90% of all the commercially available drugs that are concerned with the human mental state are amines. And a goodly number of these are primary or secondary amines. And each and every one of these could be converted to its N-hydroxyl analogue, effectively by-passing the patent protection that the originating corporation so carefully crafted. An example, just for fun. A run-away best seller right now is an antidepressant called fluoxetine, with the trade name Prozac. I will make a small wager that if I were to synthesize and taste N-hydroxy-N-methyl-3-phenyl-3-((a,a,a-trifluoro-p-tolyl)oxy)propylamine, I would find it to be an active antidepressant. Remember, Mr. Eli Lilly and Company; you read about it first, right here°
Of course, I was asked, why call it FLEA? The origin was in a classic bit of poetry. A commonly used code name for MDMA was ADAM, and I had tried making several modest modifications of the MDMA structure in the search for another compound that would maintain its particular music without the annoying tooth-grinding and occasional nystagmus, or eye-wiggle, that some users have mentioned. One of these was the 6-methyl homologue which was, with some perverse logic, called MADAM. And, following this pattern, the 6-fluoroanalogue was to be FLADAM. So, with the N-hydroxy analogue, what about HADAM? Which brought to mind the classic description of Adam's earliest complaint, an infestation of fleas. The poem was short and direct. "Adam had 'em. So, in place of HAD 'EM, the term FLEA jumped into being.
#82 G-3; 2,5-DIMETHOXY-3,4-(TRIMETHYLENE)AMPHETAMINE; 5-(2-AMINOPROPYL)-4,7-DIMETHOXYINDANE
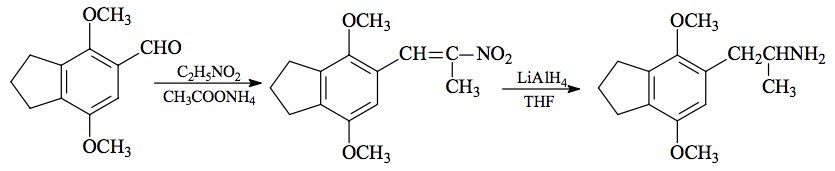

SYNTHESIS: A solution of 3.7 g of 2,5-dimethoxy-3,4-(trimethylene)benzaldehyde (see preparation under 2C-G-3) in 15 mL nitroethane was treated with 0.7 g anhydrous ammonium acetate and heated on the steam bath for 2.5 h. The excess solvent was removed under vacuum leaving some 5 mL of a deep orange-red oil which on cooling, spontaneously crystallized. This was finely ground under 10 mL MeOH, filtered, washed sparingly with MeOH, and air dried to give 3.6 g of orange crystals with a strong smell of old acetamide. The mp was 92-93 °C. All was recrystallized from 30 mL boiling MeOH to give, after filtering and drying, 2.9 g of 1-(2,5-dimethoxy-3,4-(trimethylene)phenyl)-2-nitropropene as yellow crystals with a mp of 93-94 °C. Anal. (C14H17NO4) C,H,N.
Fifty milliliters of 1 M LAH in THF was placed in an inert atmosphere, well stirred, and cooled to 0 °C with an external ice-bath. There was added, dropwise, 1.35 mL of 100% H2SO4 at a rate slow enough to minimize charring. There was then added, dropwise, 2.8 g 1-(2,5-dimethoxy-3,4-(trimethylene)phenyl)-2-nitropropene in 15 mL THF. At the end of the addition, the stirring was continued for an additional 0.5 h, and then the reaction mixture was held at reflux on the steam bath for another 0.5 h. After cooling again to ice-bath temperature, the excess hydride was destroyed with the addition of 11 mL IPA, followed by 5.5 mL 5% NaOH which converted the inorganic mass through a cottage cheese stage into a loose, filterable texture. The solids were removed by filtration, washed with additional THF, and the combined filtrates and washes stripped of solvent under vacuum. There was obtained 2.51 g of a white oil that was distilled at 115-135 °C at 0.2 mm/Hg to give 1.83 g of a clear colorless oil. This was dissolved in 8 mL IPA, neutralized with 28 drops of concentrated HCl, and diluted with 140 mL anhydrous Et2O. In about 0.5 h there started a slow snowfall of fine fluffy white crystals which was allowed to continue until no additional crystals appeared. After filtering, Et2O washing and air drying, there was obtained 1.81 g of 2,5-dimethoxy-3,4-(trimethylene)amphetamine hydrochloride (G-3) with a mp of 157-159 °C. Anal. (C14H22ClNO2) C,H.
DOSAGE: 12 - 18 mg.
DURATION: 8 - 12 h.
QUALITATIVE COMMENTS: (with 12 mg) There was a warmth, a mellowness, as things developed. No body disturbance at all, but then there were no visuals either which, for me on this particular occasion, was disappointing. The day was consumed in reading, and I identified completely with the character of my fictional hero. It was a different form of fantasy. I think I prefer music as a structural basis for fantasy.
(with 18 mg) I am at a plus three, but I am not at all sure of why it is a plus three. With my eyes closed, there are puffy clouds, but no drama at all. Music was not exciting. There could well have been easy eroticism, but there was no push in that direction. No great amount of appetite. Not much of anything, and still a plus three. Simply lying still and surveying the body rather than the visual scene gave some suggestions of neurological sensitivity, but with getting up and moving about and doing things, all was fine. The next morning I was perhaps moving a bit more slowly than usual. I am not sure that there would be reward in going higher.
EXTENSIONS AND COMMENTARY: In a comparison between the 2-carbon compound (2C-G-3) and the 3-carbon compound (G-3) the vote goes towards the phenethylamine (the 2-carbon compound). With the first member of this series (2C-G versus GANESHA) this was a stand-off, both as to quantitative effects (potency) and qualitative effects (nature of activity). Here, with the somewhat bulkier group located at the definitive 3,4-positions, the nod is to the shorter chain, for the first time ever. The potency differences are small, and maybe the amphetamine is still a bit more potent. But there are hints of discomfort with this latter compound that seem to be absent with the phenethylamine. The more highly substituted compounds (q.v.) more clearly define these differences.
#83 G-4; 2,5-DIMETHOXY-3,4-(TETRAMETHYLENE)AMPHETAMINE; 6-(2-AMINOPROPYL)-5,8-DIMETHOXYTETRALIN
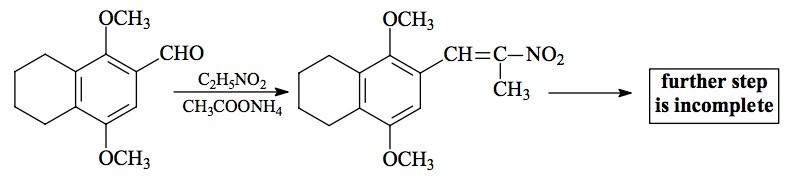

SYNTHESIS: A solution of 1,4-dimethoxy-5,6,7,8-tetrahydro-'-naphthaldehyde (see preparation under 2C-G-4) in 20 mL nitroethane was treated with 0.13 g anhydrous ammonium acetate and heated on the steam bath overnight. The volatiles were removed under vacuum and the residue, on cooling, spontaneously crystallized. This crude rust-colored product (1.98 g) was recrystallized from 15 mL boiling MeOH yielding, after filtering and air drying to constant weight, 1.33 g of 1-(2,5-dimethoxy-3,4-(tetramethylene)phenyl)-2-nitropropene as dull gold-colored crystals. The mp was 94-94.5 °C. Anal. (C15H19NO4) C,H.
DOSAGE: unknown.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: The discussion that appeared in the commentary section under 2C-G-4 applies here as well. The major struggles were in the preparation of the aldehyde itself. And although the final product has not yet been made, this last synthetic step should be, as Bobby Fischer once said in his analysis of a master's chess game following a blunder by his opponent, simply a matter of technique.
As with the phenethylamine counterpart, G-4 has a structure that lies intermediate between G-3 and G-5, both potent compounds. It is axiomatic that it too will be a potent thing, and all that now needs be done is to complete its synthesis and taste it.
#84 G-5; 3,6-DIMETHOXY-4-(2-AMINOPROPYL)BENZONORBORNANE
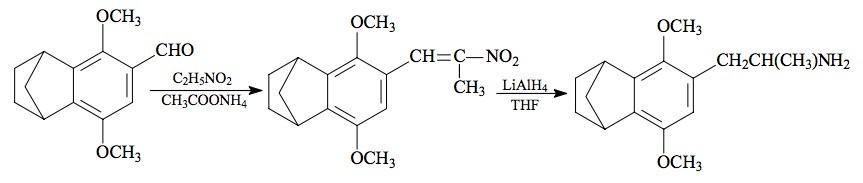

SYNTHESIS: A solution of 3.70 g 3,6-dimethoxy-4-formylbenzonorbornane (see under 2C-G-5 for its preparation) in 20 g nitroethane was treated with 0.88 g anhydrous ammonium acetate and held at steam bath temperature overnight. The excess solvent and reagent was removed under vacuum to yield a residual yellow oil. This was allowed to stand at ambient temperature for a period of time (about 3 years) by which time there was a spontaneous crystallization. The dull yellow crystals were removed by filtration and, after air drying, weighed 4.28 g. A small sample was recrystallized repeatedly from MeOH to provide a pale yellow analytical sample of 3,6-dimethoxy-4-(2-nitropropenyl)benzonorbornane with a mp of 90-91 °C. Anal. (C16H19NO4) C,H.
A solution of LAH (50 mL of 1 M solution in THF) was cooled, under He, to 0 °C with an external ice bath. With good stirring there was added 1.32 mL 100% H2SO4 dropwise, to minimize charring. This was followed by the addition of 4.1 g 3,6-dimethoxy-4-(2-nitropropenyl)benzonorbornane in 20 mL anhydrous THF over the course of 10 min. The reaction mixture was stirred and brought to room temperature over the course of 1 h. This was then brought to a gentle reflux on the steam bath for 0.5 h, and then all was cooled again to 0 °C. The excess hydride was destroyed by the cautious addition of 10 mL IPA followed by 5 mL 5% NaOH and sufficient H2O to give a white granular character to the oxides. The reaction mixture was filtered, and the filter cake washed with THF. The filtrate was stripped of solvent under vacuum providing a pale amber oil that was distilled at 125-140 °C at 0.2 mm/Hg to give 2.5 g of an almost white oil. This was dissolved in 10 mL IPA, neutralized with 25 drops of concentrated HCl, and then diluted with 140 mL anhydrous Et2O. There appeared, after about two minutes, white crystals of 3,6-dimethoxy-4-(2-aminopropyl)benzonorbornane hydrochloride (G-5) which, after filtration and air drying, weighed 2.47 g.
DOSAGE: 14 - 20 mg.
DURATION: 16 - 30 h.
QUALITATIVE COMMENTS: (with 15 mg) As part of the audience at the San Francisco conference, Angels, Aliens and Archtypes, I could simply listen and observe without having to participate. Each speaker stood in a cone of light that was beautifully bright and colorful, casting everything else on the stage into obscurity. Maybe angels really are illuminated from above, and the aliens lurk out of sight until it is their turn. Where does one look for the archetypes? A half of a cream cheese sandwich was all I could eat, and even at dinner that evening I was not hungry. Sleep that evening was difficult.
(with 20 mg) Very slow to come on, but then it was up there all of a sudden. There is an unexpected absence of visual activity despite being at a full +++. The mental activity is excellent, with easy writing and a positive flow of ideas. But an absence of the bells and whistles that are expected with a psychedelic in full bloom. There is a real drop by the 16th hour and the next day was free of effect except for occasional cat-naps.
(with 20 mg) The transition period, which usually lasts for most compounds for the first hour or two, with this seems to be much longer. This presages a long-acting material, as usually the slow-in slow-out rule applies. But there are exceptions. There is an indifference towards the erotic, but no separation at all from personal interactions and emotions. I believe in integration, not separation of all parts of ourselves, distrusting any drug states (particularly those that have the reputation of being strongly Tcosmic)' which divorce the consciousness from the body. And with this material there is no separation from feelings, only from my particular color language.
EXTENSIONS AND COMMENTARY: This is as potent as any of the three-carbon Ganesha compounds, but it somehow lacks a little something that would have made it a completely favorite winner. Perhaps it is the generally commented upon absence of visual and related sensory entertainment. There seems to be no bodily threat to discourage further exploration, but there simply was not the drive to explore it much. The comments concerning the enlargement of the ring system (mentioned under 2C-G-5) are equally valid here. The RshrubberyS that is the hallmark of the Ganesha family is, with G-5, about as bulky as has ever been put onto a centrally active molecule. The norbornane group has a one carbon bridge and a two carbon bridge sticking out of it at odd angles. The replacement of the one-carbon bridge with a second two-carbon bridge would make the compound G-6. It would be makeable, but is there really a driving reason to do so? There is a simplification intrinsic in this, in that G-5 actually has two centers of asymmetry (the a-carbon atom on the amphetamine chain, and the norbornyl area itself) and so it is really a mixture of two racemic diastereoisomers. G-6 would still be a racemate, but it would be only a single compound, as are all the other substituted amphetamine derivatives.
Someday I may try making G-6, but it's not a high priority right now.
#85 GANESHA; G; 2,5-DIMETHOXY-3,4-DIMETHYLAMPHETAMINE
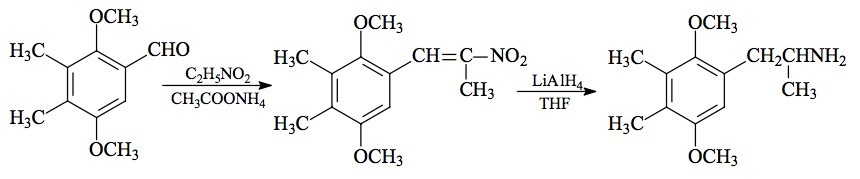

SYNTHESIS: A solution of 15.4 g 2,5-dimethoxy-3,4-dimethylbenzaldehyde (see under 2C-G for the preparation) in 50 mL nitroethane was treated with 3 g anhydrous ammonium acetate and heated on the steam bath for 12 h. The excess nitroethane was removed under vacuum, and the residual oil was diluted with a equal volume of MeOH. There was the slow generation of deep red cottage-cheese-like crystals which were removed by filtration and air-dried to constant weight (9.3 g) with a mp 71-74 °C. Recrystal-lization from MeOH (10 ml/g) gave an analytical sample of 1-(2,5-dimethoxy-3,4-dimethylphenyl)-2-nitropropene with a mp of 82 °C sharp. Anal. (C13H17NO4) C,H,N. The NMR spectra (in CDCl3) and CI mass spectrograph (MH+ = 252) were proper.
To a suspension of 3.3 g LAH in 200 mL refluxing THF, well stirred and maintained under an inert atmosphere, there was added 4.2 g 1-(2,5-dimethoxy-3,4-dimethylphenyl)-2-nitropropene in 25 mL THF. The mixture was held at reflux for 48 h. After cooling, 3.3 mL H2O was added cautiously to decompose the excess hydride, followed by 3.3 mL 15% NaOH and finally another 10 mL H2O. The inorganic solids were removed by filtration, and washed with additional THF. The combined filtrate and washes were stripped of solvent under vacuum, and the residue (4.7 g of a deep amber oil) dissolved in dilute HCl. This was washed with CH2Cl2 (3x75 mL), then made basic with 5% NaOH and extracted with CH2Cl2. Removal of the solvent under vacuum yielded an amber oil that was distilled (105-115 °C at 0.4 mm/Hg) to give 1.2 g of a white oil. This was dissolved in 8 mL IPA, neutralized with 15 drops of concentrated HCl, and diluted with 250 mL anhydrous Et2O. After a period of time, there was a spontaneous appearance of white crystals which were removed by filtration, Et2O washed, and air dried. Thus was obtained 1.0 g of 2,5-dimethoxy-3,4-dimethylamphetamine hydrochloride (GANESHA) with a mp of 168-169 °C. This was not improved by recrystallization from either EtOAc or nitroethane. Anal. (C13H22ClNO2) N.
DOSAGE: 20 - 32 mg.
DURATION: 18 - 24 h.
QUALITATIVE COMMENTS: (with 24 mg) There was a slow buildup to a ++ or more over the course of about three hours. Extremely tranquil, and no hint of any body toxicity whatsoever. More than tranquil, I was completely at peace, in a beautiful, benign, and placid place. There was something residual that extended into the sleep period, and was possibly still there in the morning. Probably I was simply tired from an inadequate sleep.
(with 32 mg) A rapid and full development. Lying down with music, the eyes-closed visuals were quite something. There was sudden awareness of a potential toe cramp which I possibly exaggerated, but it kept spinning itself into my awareness, and somehow locked in with my visual imagery. It was not easy to keep the visual/somatic/ cognitive worlds in their proper places. The almost-cramp went away and I forgot about it. There was a back spasm somewhere in this drama, and it really didn't matter either. This dosage may be a bit much for good housekeeping, though° Towards the end of the experiment, I looked at a collection of photos from a recent trip to Europe, and the visual enhancement was wonderful. A rolling +++.
EXTENSIONS AND COMMENTARY: This compound was the seventh of the ten possible Classic Ladies. I have mentioned the concept already under the discussions on ARIADNE. This is the teutonic replacement of each of the distinguishable hydrogen atoms of DOM with a methyl group. The findings with GANESHA were a total surprise. The extension of a hydrogen in the 3-position of DOM with a methyl group should have a minor influence on its steric association with whatever receptor site might be involved. A much greater impact might come not from the size of the group but from its location. This, coupled with a full order of magnitude of decrease in potency, seemed to call for an involvement of that particular position as being one that is affected by metabolism. And since the activity is decreased, the obvious role is in the blocking of the metabolic promotion of DOM-like things to active intermediates.
The remarkable point being emphasized here is that the placement of a dull methyl group at a dull position of the DOM molecule actually inactivated (for all intents and purposes) the activity of DOM. It is not the presence of the methyl that has decimated the potency, but the removal of the hydrogen atom.
How can such a hypothesis be explored? A historic premise of the medicinal chemist is that if a structure gives an unusual response in a receptor, vary it slightly and see how the response varies. This is exactly the principle that led to the ten Classic Ladies, and with this particular Lady (who actually turned out to be a gentleman), the same concept should hold. There are two involved methyl groups in GANESHA, one at the 3-position and one at the 4-position. Why not homologate each to an ethyl group, and as a wrap up make both of them into ethyl groups. Look at the differences along two lines of variation; the effects of the homologation of the 3- and 4-positions, coupled with the effects of the homologation intrinsic in the comparison of the two-carbon chain of the phenethylamine with the three-carbon chain of the amphetamine.
There are thus six compounds involved in such a study. And they have been named (as have all the other GANESHA analogues) in accordance with the collective carbon inventory in and about these two ring positions. The first two compounds are related to DOET and to 2C-E. Maintain the methyl group at the 3-position but homologate the 4-position to an ethyl. The ring pattern would become 2,5-dimethoxy-4-ethyl-3-methyl, and the phenethylamine and amphetamine would be called 2C-G-12 and G-12 respectively (a one carbon thing, the methyl, at position-3 and a two carbon thing, an ethyl, at position-4). Reversal of these groups, the 3-ethyl homologues of 2C-D and DOM would thus become 2C-G-21 and G-21. And, finally, the diethyl homologues would be 2C-G-22 and G-22. In each of these cases, the paired numbers give the lengths of the chains at the two positions, the 3- and the 4-positions that are part of the GANESHA concept. And this code is easily expandable to longer things such as 2C-G-31 and 2C-G-41, which would be the 3-propyl-4-methyl, and the 3-butyl-4-methyl homologues, resp.
Unfortunately, these six initially proposed compounds have so far resisted all logical approaches to synthesis, and are at present still unknown. What has been successfully achieved, the building up of a big bulky hydrocarbon glob at these positions, has rather unexpectedly led to a remarkable enhancement of potency. As with all true exploration into areas of the unknown, the deeper you get, the less you understand.
#86 G-N; 1,4-DIMETHOXYNAPHTHYL-2-ISOPROPYLAMINE
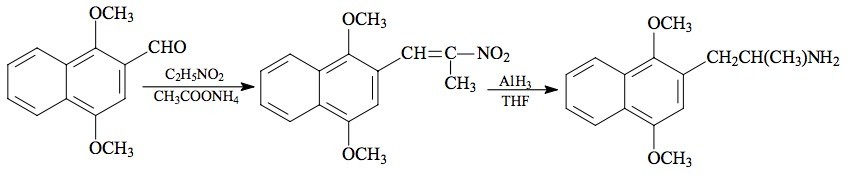

SYNTHESIS: To a solution of 3.9 g 1,4-dimethoxy-2-naphthaldehyde (see under 2C-G-N for the preparation) in 13.5 mL nitroethane there was added 0.7 g anhydrous ammonium acetate, and the mixture heated on the steam bath for 5 h. The deep orange reaction mixture was stripped of excess solvent under vacuum. The residue was a red oil that, upon dilution with two volumes MeOH, immediately set to orange crystals. This crude product (mp 115-118 °C) was recrystallized from 70 mL EtOH to yield, after filtering and air drying, 3.3 g of 1-(1,4-dimethoxy-2-naphthyl)-2-nitropropene as gold-orange crystals, with a mp of 121-123 °C. Recrystallization from MeOH gave a gold-colored product with a mp of 119-120 °C. Anal. (C15H15NO4) C,H,N.
A solution of LAH (50 mL of 1 M solution in THF) was cooled, under He, to 0 °C with an external ice-bath. With good stirring there was added 1.32 mL 100% H2SO4 dropwise, to minimize charring. This was followed by the addition of 3.12 g 1-(1,4-dimethoxy-2-naphthyl)-2-nitropropene in 40 mL anhydrous THF. After stirring for 1 h, the temperature was brought up to a gentle reflux on the steam bath for 0.5 h, and then all was cooled again to 0 °C. The excess hydride was destroyed by the cautious addition of 16 mL IPA followed by 6 mL 5% NaOH to give a white, filterable, granular character to the oxides, and to assure that the reaction mixture was basic. The reaction mixture was filtered, and the filter cake washed with additional THF. The combined filtrate and washes were stripped of solvent under vacuum providing 3.17 g of a deep amber oil. Without any further purification, this was distilled at 140-160 °C at 0.3 mm/Hg to give 1.25 g of a pale yellow oil. This was dissolved in 8 mL IPA, neutralized with 20 drops of concentrated HCl, and diluted with 60 mL anhydrous Et2O which was the point at which the solution became slightly turbid. After a few min, fine white crystals began to form, and these were eventually removed, washed with Et2O, and air dried to provide 1.28 g 1,4-dimethoxynaphthyl-2-isopropylamine hydrochloride (G-N) as the monohydrate salt. The mp was 205-206 °C. Even after 24 h drying at 100 °C under vacuum, the hydrate salt remained intact. Anal. (C15H20ClNO2aH2O) C,H.
DOSAGE: unknown.
DURATION: unknown,
EXTENTIONS AND COMMENTARY: The evaluation of this compound is not yet complete. An initial trial at the 2 milligram level showed neither central action, nor toxicity. It could be guessed from the activity of the two-carbon counterpart, that an active level will be found in the tens of milligrams area. But, as of the moment, this level is not known to anyone, anywhere, because no one has yet defined it. And when the potency is finally found out, the nature of the activity will also have been found out, all the result of a magical interaction of a virgin compound with a virgin psyche. At the immediate moment, the nature of G-N is not only unknown, it has not yet even been sculpted. There can be no more exciting area of research than this, anywhere in the sentient world.
#87 HOT-2; 2,5-DIMETHOXY-4-ETHYLTHIO-N-HYDROXYPHENETHYLAMINE
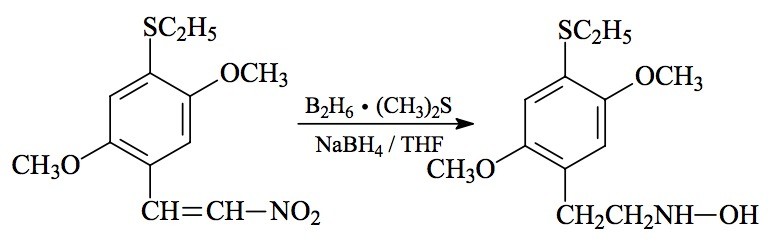

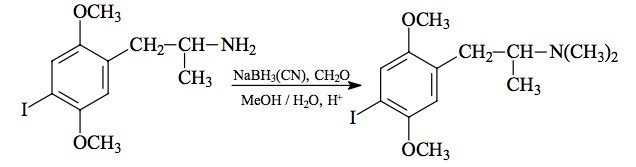
SYNTHESIS: A solution of 5.50 g 2,5-dimethoxy-4-ethylthio-'-nitrostyrene (see under 2C-T-2 for its preparation) was made in 80 mL boiling anhydrous THF. On cooling, there was some separation of a fine crystalline phase, which was kept dispersed by continuous stirring. Under an inert atmosphere there was added 3.5 mL of a 10 M borane dimethylsulfide complex, followed by 0.5 g sodium borohydride as a solid. There was a slight exothermic response, and the color slowly faded. Stirring was continued for a week. There was then added 40 mL H2O and 20 mL concentrated HCl, and the reaction mixture heated on the steam bath for 15 minutes, with the THF at reflux. After cooling again to room temperature, all was poured into 1 L H2O and washed with 3x75 mL CH2Cl2, which removed all of the color but little of the product. The aqueous phase was made basic with 25% NaOH, and extracted with 3x75 mL CH2Cl2. The extracts were pooled and the solvent removed under vacuum to give a residue of 3.88 g of an amber oil. This was dissolved in 30 mL IPA, acidified with concentrated HCL to a bright red on universal pH paper, and then diluted with 200 mL anhydrous Et2O. After a short period of time, crystals started to form. These were removed by filtration, washed with Et2O, and air dried to constant weight. Thus was obtained 2.86 g 2,5-dimethoxy-4-ethylthio-N-hydroxyphenethylamine hydrochloride (HOT-2) as off-white crystals, with a melting point of 122 °C with decomposition. Anal. (C12H20ClNO3 S) H; C: calcd, 49.05; found, 50.15, 49.90.
DOSAGE: 10 - 18 mg.
DURATION: 6 - 10 h.
QUALITATIVE COMMENTS: (with 12 mg) Tastes OK. Some activity noticed in 30 minutes. Very smooth rise with no body load for next two hours. At that time I noted some visuals. Very pleasant. The bright spots in the painting over the fireplace seemed to be moving backwards (as if the clouds were moving in the painting). Upon concentrating on any item, there was perceptual movement with a little flowing aspect. The visuals were never all that strong, but could not be turned off during the peak. At hour three there was still some shimmering, and it was hard to focus when reading. Additionally, there was difficulty concentrating (some mental confusion). The material seemed to allow erotic actions; there was no problem about obtaining an erection. I ate very well, some crazy dips, as well as a fabulous cake. A very gentle down trend and I became close to baseline by 6 or 7 PM. I had no trouble driving. The dosage was good for me. I did not want more or less.
(with 12 mg) Comes on smoothly, nicely. In 40 minutes I feel nice euphoria, feel home again. Then I begin to get uncomfortable feelings. Gets more and more uncomfortable, feel I am sitting on a big problem. Blood pressure, pulse, go up considerably. Have hard time communicating, lie down for a while, get insight that most important thing for me to do is learn to listen, pay attention to what is going on. I do this the rest of the day, at first with considerable difficulty, then easier and easier. Discomfort stays with me for several hours, and although I get more comfortable towards the end of the day, I am never animated or euphoric. I feel very humbled, that I have a great deal to work out in my life. The next day I find myself very strong and empowered. I see that all I have to do is let things be as they are° This feels marvelous, and a whole new way to be Q much more relaxed, accepting, being in the moment. No more axes to grind. I can be free.
(with 18 mg) I found myself with complete energy. I was completely centered with an absolute minimum of the dark edges that so often appear as components of these experiences. The ease of talking was remarkable. There was some blood-pressure run-up in the early part of the day, but that quickly returned to normal. I would repeat without hesitation.
EXTENSIONS AND COMMENTARY: Again, a case of where the potency range of the Rhot,S or hydroxylated compound (HOT-2, 10 to 18 milligrams) is very similar to that of the non-hydroxylated prototype (2C-T-2, 12-25 milligrams). It seems to be a well tolerated, and generally pleasant material, with a mixture of sensory as well as insightful aspects. Something for everyone.
#88 HOT-7; 2,5-DIMETHOXY-N-HYDROXY-4-(n)-PROPYLTHIOPHENETHYLAMINE
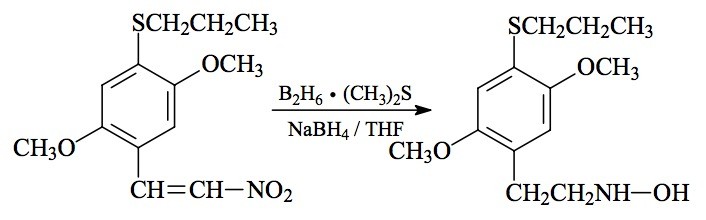

SYNTHESIS: A well-stirred solution of 1.77 g 2,5-dimethoxy-'-nitro-4-(n-propylthio)styrene (see under 2C-T-7 for its preparation) in 20 mL anhydrous THF was placed in an He atmosphere and treated with 1.5 mL of 10 M borane-dimethyl sulfide complex. This was followed by the addition of 0.2 g sodium borohydride, and the stirring was continued at room temperature for a week. The volatiles were removed under vacuum, and the residue was treated with 20 mL dilute HCl and heated on the steam bath for 30 min. The cooled yellow solution set up as solids. The addition of H2O was followed by sufficient K2CO3 to make the aqueous phase basic. All efforts to work with an acidified aqueous phase resulted in terrible emulsions. The basic phase was extracted with 3x75 mL CH2Cl2, and the pooled extracts washed with H2O, then stripped of solvent under vacuum. The residual yellow oil was dissolved in 20 mL IPA, neutralized with 15 drops of concentrated HCl, and then diluted with 50 mL anhydrous Et2O. After a few minutes stirring, a white crystalline solid separated. This was removed by filtration, washed with Et2O, and air dried to constant weight to provide 0.83 g of 2,5-dimethoxy-N-hydroxy-4-(n)-propylthiophenethylamine hydrochloride (HOT-7).
DOSAGE: 15 - 25 mg.
DURATION: 6 - 8 h.
QUALITATIVE COMMENTS: (with 15 mg) I am lightheaded, and maybe a little tipsy. I am well centered, but I don't want to go outside and meet people. Shades of alcohol woozy. The effects were going already by the fifth hour and were gone by the seventh hour. I would call it smoothly stoning.
(with 22 mg) The transition into the effects was a bit difficult, with a faint awareness in the tummy. But by the second hour it was quite psychedelic, and the body was not thought of again, except in terms of sexual fooling around. Very rich in eyes-closed imagery, and very good for interpretive and conceptual thinking. But the eyes-open visuals were not as much as they might have been. At the seventh hour, drifted into an easy sleep.
(with 22 mg) The experience was very positive, but at each turn there seemed to be a bit of sadness. Was it a complete plus three experience? Not quite. But it didn't miss by much. The erotic explorations somehow just failed to knit by the thinnest of margins. It was a truly almost-magnificent experience.
EXTENSIONS AND COMMENTARY: There is a working hypothesis that has been growing in substance over the last few years in this strange and marvelous area of psychedelic drugs. It all was an outgrowth of the rather remarkable coincidence that I had mentioned in the discussion that followed MDOH. There, an assay of what was thought to be MDOH gave a measure of activity that was substantially identical to MDA, and it was later found out that the material had decomposed to form MDA. So, MDA was in essence rediscovered. But when the true, valid, and undecomposed sample of MDOH was actually in hand, and assayed in its own rights, it was found to have a potency that really was the same as MDA. So, the working hypothesis goes something like this:
AN N-HYDROXY AMINE HAS APPROXIMATELY THE SAME POTENCY AND THE SAME ACTION AS ITS N-HYDROGEN COUNTERPART.
Maybe the N-hydroxy compound reduces to the N-H material in the body, and the latter is the intrinsically active agent. Maybe the N-H material oxidizes to the N-hydroxy material in the body, and the latter is the intrinsically active agent. Either direction is reasonable, and there is precedent for each. The equivalence of MDA and MDOH was the first suggestion of this. And I have made a number of NH vs. NOH challenges of this hypothesis. The interesting 2C-T-X series has provided a number of amines that are amenable to N-hydroxylation, and this is the first of them. And, after all, if you put a hydroxy (HO) group on a thio material (T), you have a HOT compound.
So, as far as nomenclature is concerned, the family of N-hydroxy analogues of N-H amines is known as the HOT family.
How does HOT-7 compare with 2C-T-7? They are almost identical. The same range of dose (centering on 20 milligrams) and if anything, perhaps slightly less long lived. Lets try some other N-hydroxys°
#89 HOT-17; 2,5-DIMETHOXY-4-(s)-BUTYLTHIO-N-HYDROXYPHENETHYLAMINE
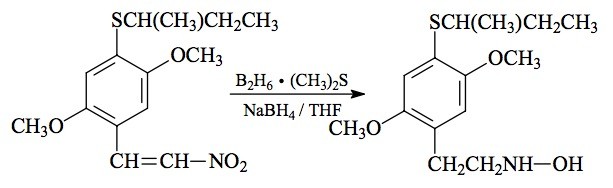

SYNTHESIS: To a well-stirred solution of 6.08 g 2,5-dimethoxy-4-(s)-butylthio-'-nitrostyrene (see under 2C-T-17 for its preparation) in 80 mL anhydrous THF under a He atmosphere, there was added 3.5 mL 10 M borane dimethylsulfide complex, followed by 0.5 g of sodium borohydride. As the stirring continued, the slightly exothermic reaction slowly faded from bright yellow to pale yellow, and eventually (after three days stirring) it was substantially colorless. There was then added 80 mL of 3 N HCl and the mixture heated on the steam bath for 1 h, and then allowed to return to room temperature. An additional 600 mL H2O was added (there was a combination of crystals and globby chunks in the aqueous phase) and this was then extracted with 3x75 mL CH2Cl2. The color went completely into the organic phase. This was washed with 2x50 mL aqueous K2CO3, yielding a rusty-red colored CH2Cl2 solution, which on removal of the solvent, yielded 4.5 g of a red oil. A side effort to make the sulfate salt at this stage with H2O and a little H2SO4, indeed gave solids, but all of the color remained in the sulfate salt. The red oil was dissolved in 45 mL IPA and neutralized with concentrated HCl to bright red, not yellow, on universal pH paper. The addition of 350 mL anhydrous Et2O instituted the slow precipitation of white crystals. After filtering and air drying, there was obtained 1.32 g 2,5-dimethoxy-4-(s)-butylthio-N-hydroxyphenethylamine hydrochloride (HOT-17). The aqueous phase from above was just neutralized with 25% NaOH (cloudy, slightly pink color) and then made basic with K2CO3 (the color becomes green). This was extracted with 3x75 mL CH2Cl2, the extracts pooled, and the solvent removed to yield 0.5 g of a white oil. This was dissolved in 5 mL IPA, neutralized with concentrated HCl, and diluted with a equal volume of Et2O. An additional 0.36 g of product was thus obtained.
DOSAGE: 70 - 120 mg.
DURATION : 12 - 18 h.
QUALITATIVE COMMENTS: (with 70 mg) There was a light feeling, a little off-the-ground feeling, which made walking about a most pleasant experience. No distortion of the senses. And there was no sense of the beginning of a drop of any kind until about the eighth hour. Sleeping was a bit tricky but it worked out OK (at the twelfth hour of the experience). A completely valid ++.
(with 120 mg) HOT-17 has an unbelievably GRIM taste Q not bitter, but simply evil. There is a steady and inexorable climb for three hours to a sound and rolling plus three. There was absolutely no body difficulty, but there was still something going on upstairs well into the next day. Writing was surprisingly easy; I was completely content with the day, and would be interested in exploring it under a variety of circumstances.
(with 120 mg) This is my first time with this material. It is 4:45 PM. Small nudge at 30 minutes, but not too real. At one hour, threshold, quite real. 6:15 to a +1. By 7:25, +3 about. 7:45, no doubt +3. Possibly still climbing; I hope so. No body discomfort at all, no apparent body push. This aspect of it is similar to the easy body of the HOT-2. However, it's at times like these that I reflect on just exactly how hard-headed we two are. I mean, +3 is no longer the out-of-body, nearly loss of center state it used to be, four years ago. The question intrudes: would a novice experience this as a very scary, ego-disintegrating kind of experiment, or not? Silly question which answers itself. Yes, of course. At 3 hours, aware of some mild time-distortion. More a tendency to not think in terms of clock-time, than actual distortion. The mind lazy when attempting to keep track of clock time. Feel it would be quite easy and pleasant to continue writing. The energy could very well go in that direction. However, the idea of the erotic is also quite agreeable. This is, so far, a good-humored Buddha area of the self.
EXTENSIONS AND COMMENTARY: Two virtues sought by some users of psychedelic drugs are high intensity and brief action. They want a quicky. Something that is really effective for a short period of time, then lets you quickly return to baseline, and presumably back to the real world out there.
Intensity is often (but not always) regulated by dose. The pharmacological property of dose-dependency applies to many of these drugs, in that the more you take, the more you get. If you want more intensity, take a second pill. And often, you get a longer duration as an added property. But it is instructive to inquire into the rationale that promotes brevity as a virtue. I believe that it says something concerning the reasons for using a psychedelic drug. A trade off between learning and entertainment. Or between the achieving of something and the appearance of achieving something. Or, in the concepts of the classics, between substance and image.
In a word, many people truly believe that they cannot afford the time or energy required for a deep search into themselves. One has to make a living, one has to maintain a social life, one has a multitude of obligations that truly consume the oh-so-few hours in the day. I simply cannot afford to take a day off just to indulge myself in such-and-such (choose one: digging to the bottom of a complex concept, giving my energies to those whom I can help, to search out my inner strengths and weaknesses) so instead I shall simply do such-and-such (choose one: read the book review, go to church on Sunday morning, use a short-acting psychedelic). The world is too much with us. This may be a bit harsh, but there is some merit to it.
HOT-17 is by no means a particularly potent compound. The hundred milligram area actually has been the kiss of death to several materials, as it is often at these levels that some physical concerns become evident. And it certainly is not a short lived compound. But, as has been so often the case, the long lived materials have proven to be the most memorable, in that once the entertainment aspect of the experience is past you, there is time for dipping deeply into the rich areas of the thought process, and the working through of ideas and concepts that are easily available. And when this access is coupled to the capability of talking and writing, then a rewarding experience is often the result.
As with the parent compound, 2C-T-17 itself, the presence of an asymmetric carbon atom out there on the (s)-butyl side chain will allow the separation of HOT-17 into two components which will be different and distinct in their actions. The activity of the racemic mixture often is an amalgamation of both sets of properties, and the separate assay of each component can often result in a fascinating and unexpected fractionation of these properties.